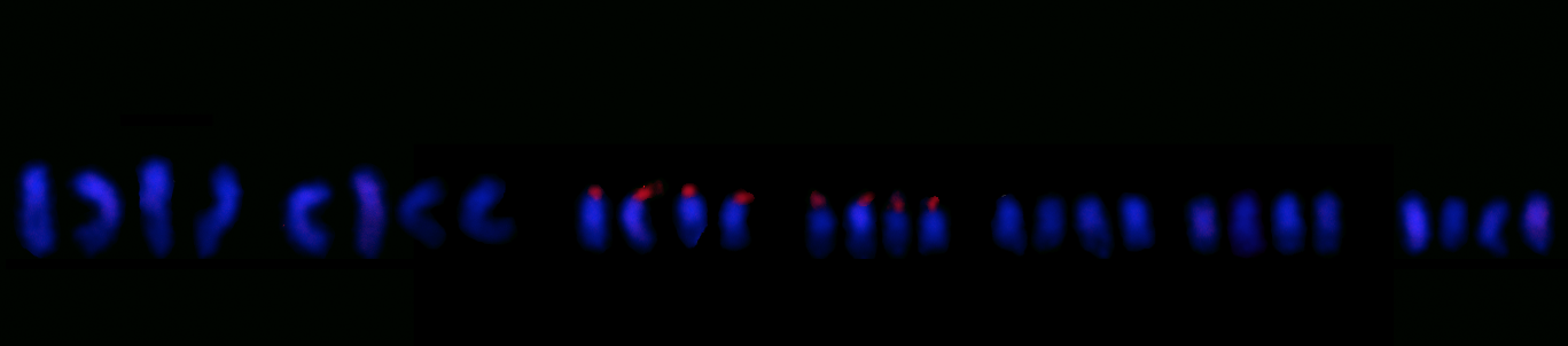
Current Research

Our research interests are in the evolutionary role of polyploidy, or whole-genome duplication, and how it functions as a speciation mechanism. We investigate the impact of polyploidy in flowering plants using genomic and bioinformatics methods, while drawing from the fields of physiology and ecology to place the findings into an evolutionary and ecological context.


The genomic consequences of polyploidy ultimately stem from gene dosage effects incurred by a doubling of every copy of every gene in the genome. Our research explores how polyploidy-driven shifts in gene dosage impact gene expression.
We are particularly interested in how patterns of gene expression in polyploids change over time as physiological stress is imposed. One hypothesis is that in times of high stress, polyploids are able to exhibit a transgressive response transcriptomically due to the maintenance of additional gene copies. In other words, a tetraploid with four genomic copies may be able to up-regulate its stress response to a greater degree than the diploid with two genomic copies.
RNAseq normalization
Current RNAseq-based methods for comparing transcript abundances rest on the assumption that total transcriptome size does not vary between groups. Due to this limitation, previous studies of polyploid gene expression have been limited to comparisons of transcript concentration relative to the transcriptome, analogous to molarity, rather than transcript abundance.

To better investigate polyploid gene expression, we have adapted the use of synthetic RNAs to enable simultaneous comparisons of transcript abundance per cell and transcript abundance per biomass, independently from transcriptome size. Our approach was applied on a transcriptome-wide basis using diploid and autopolyploid Tolmiea (Saxifragaceae) as a natural model system. We are currently interested in expanding the use of this method to investigations into rates of translation, and also application in non-polyploid systems.
Polyploidy and translation
A largely overlooked step in the path from genotype to phenotype following polyploidy is the translation of mRNA to protein. The global pool of mRNA under active translation, the translatome, serves as an intermediary between the transcriptome and the proteome. The translatome is one of the most promising places to begin exploring the commonly observed discontinuity between changes in the transcriptome and the proteome. With polyploidy leading to an increased production of mRNAs, it is critical to investigate whether they also have an increased capacity for translation.
In many cases, polyploidy increases cell volume, which may facilitate a larger pool of ribosomes to draw upon, but this has not yet been investigated. Up-regulated transcription in a polyploid may ultimately fail to have a physiological effect if the transcripts are subjected to a translational bottleneck. Using a combination of the three-normalization RNAseq method developed by our lab and high-throughput ribosome profiling, we hope to characterize ribosome activity per locus and assess whether translational throughput in polyploids scales with the increase in transcription.

The Visger lab is also actively involved in both methods development and applied efforts in ecological modeling.
Collaborations
The integrative nature of our work has led to successful collaborations with people in different departments, universities, and countries. While we intend to continue many of our current collaborations, we hope to forge new, mutually beneficial collaborations here at Sacramento State.